
Huntingtons Disease
Brief Overview
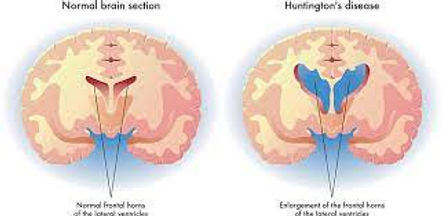
Huntington's disease is a Neurodegenerative Disease that affects the brain and central nervous system that causes unwanted movements, behavioral and psychiatric issues (Roos, 2010) (Look at image 5). It is a single gene disorder and involves the HTT gene and the Huntingtin protein on chromosome 4 (Roos, 2010; "Examples of ", 2014). A key job of this protein is in nerve cells to send signals to the brain and is an autosomal dominant trait. Symptoms of this disease include mood swings, poor memory, lack of coordination, uncontrolled walking, twitching and etc. Current treatments cannot slow or destroy the disease, but they ease the life of the person affected, and for future treatments, we hope to have this a process to replace nerve cells by transplanting stem cells(Roos, 2010).
Deeper Expanation
Huntington's disease was recognized by George Huntington in 1842 and research has been going on since then. It was not until 1993, where they found the gene and chromosome Huntington's is linked to. As mentioned in the brief overview, Huntington's is a neurodegenerative disorder that affects the central nervous system. This causes unwanted physic iatric and behavioral issues. This disease passes on from generation to generation since it follows an autosomal dominant pattern (Roos, 2010).
This disease occurs because of an extensive repeat in the CAG codon, which codes for glutamic. Huntington's disorder is a disorder that is affected by the environmental population, as Caucasian people have a higher chance to receive the disease than Japanese people. This may have been caused by diet, but this cause is still unknown. Autonomic nervous system errors, unintended weight loss, and sleep problems are just a few of the common issues people face with Huntington's. The range of people who are diagnosed with this disease ranges from 2 - 85 years, but the mean age is 30 - 50 years, and being diagnosed for about 17-20 years before death ("Examples of ", 2014).
Some motor symptoms and signs that are observable include facial expression issues, not being able to do simple things like walking talking, and even swallowing, and the presence of hypo and hypermetria are some of the common ones. This motor disturbance progress over the course of daily life and will be thee continuously when the person affected is awake. There are no observable psychiatric and behavioral signs of this disease. About 33% to 76% of the people affected have very observative psychiatric issues causing depression, weight loss, and anxiety. This destroys self-esteem, causes a loss of interest, and causes the person affected to think into more deeper and complex topics like suicide. Cognitive decline is a common sign of Huntington's are they cannot execute simple functions well. They cannot organize their life, or plan things accordingly as well enough anymore ("Examples of ", 2014).
Juvenile's Huntington's disease occurs when you are diagnosed with Huntington's under the age of 30, which occurs due to the extensive CAG codon repeat. In this case, it repeats more than 55 times, causing it to come earlier. There is also a preclinical and after clinical-stage, where preclinical is where someone in your family line was possibly diagnosed with Huntington's and you are worrying, and clinical is the diagnosis of the disease. As discussed before, Huntington's is caused by the elongated CAG repeat on chromosome 4 on exon 1 (Image 6). The wild-type is associated with 6-26 repeats, and you are diagnosed with Huntington's if you have more than 36 repeats of CAG. The longer the CAG codon repeats, the earlier the onset of the disease. You will know you have Huntington's at an earlier age if you have a lot of repeats (Image 7, 8 references this fact). The normal wild-type plays a role in synaptic function, and research shows that the mutant form can either lead to a gain or loss in function("Examples of ", 2014).
Huntington's disease does not have a proper diagnosis that has been found by current scientists, but there are many ways to test if you have this disease. Informed consent from the patient and brain MRI scans are some of the possible ways to test if you have this disease. This cause can also be caused due to iatrogenic disorders, meaning getting the disorder from a prescribed drug by a physician. This is only about 1% of the cases of Huntington's meaning it is extremely rare but not impossible. If you believe to have this disease, you can follow a certain few steps to make sure are safe. Step 1: Consultation with a clinical geneticist. Step 2: Blood sampling after a second consultation. Step 3: A consolation where disclosure is planned (Roos, 2010).
There is a prenatal procedure for this, if you believe that your child is almost guaranteed this disease, you can remove this gene. If a couple decides to do so, they can have their embryo removed and put in a lab, the Huntington's disease cell removed, and placed back to make sure they will not inherit it genetically. Since there is no possible cure at this point in time, using certain drugs is up to the physician but no drug can cure this disease and instead eases your lifestyle (Roos, 2010).
Other diseases similar to this include hyperkinesia, which can be treated using many different drugs and methods but this disease cannot. If a person in your family has this disorder, therapy is a must as they will have moments of depression and hopelessness where they will need help. Huntingtin's disease has been having research going on for more than 150 years, but there is no still cure and many lives leading to death are quite frustrating, and painful which is why something needs to be done (Roos, 2010).
Background Info:
Important Facts:
Symptoms & Signs:
Genetical & DNA Information:
Diagnosis:
Prenatal Procedure:
Other Similar Diseases:
Image 5: Basic Description of the Brain of people with and without Huntingtins
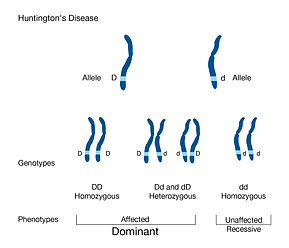
Image 6: Looking at two alleles from each parent, the genotype and if you received the disease or not.
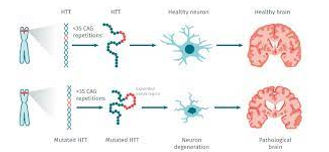
Image 7: Comparing some features of a regular person vs a Huntingtin's disease affected person

Image 8: Looking at the elongated CAG repeat, the affect on the brain and the chance of receiving the disease.